


주메뉴
- About IBS 연구원소개
-
Research Centers
연구단소개
- Research Outcomes
- Mathematics
- Physics
- Center for Underground Physics
- Center for Theoretical Physics of the Universe (Particle Theory and Cosmology Group)
- Center for Theoretical Physics of the Universe (Cosmology, Gravity and Astroparticle Physics Group)
- Dark Matter Axion Group
- Center for Artificial Low Dimensional Electronic Systems
- Center for Theoretical Physics of Complex Systems
- Center for Quantum Nanoscience
- Center for Exotic Nuclear Studies
- Center for Van der Waals Quantum Solids
- Center for Relativistic Laser Science
- Chemistry
- Life Sciences
- Earth Science
- Interdisciplinary
- Center for Neuroscience Imaging Research (Neuro Technology Group)
- Center for Neuroscience Imaging Research (Cognitive and Computational Neuroscience Group)
- Center for Algorithmic and Robotized Synthesis
- Center for Genome Engineering
- Center for Nanomedicine
- Center for Biomolecular and Cellular Structure
- Center for 2D Quantum Heterostructures
- Center for Quantum Conversion Research
- Institutes
- Korea Virus Research Institute
- News Center 뉴스 센터
- Career 인재초빙
- Living in Korea IBS School-UST
- IBS School 윤리경영

주메뉴
- About IBS
-
Research Centers
- Research Outcomes
- Mathematics
- Physics
- Center for Underground Physics
- Center for Theoretical Physics of the Universe (Particle Theory and Cosmology Group)
- Center for Theoretical Physics of the Universe (Cosmology, Gravity and Astroparticle Physics Group)
- Dark Matter Axion Group
- Center for Artificial Low Dimensional Electronic Systems
- Center for Theoretical Physics of Complex Systems
- Center for Quantum Nanoscience
- Center for Exotic Nuclear Studies
- Center for Van der Waals Quantum Solids
- Center for Relativistic Laser Science
- Chemistry
- Life Sciences
- Earth Science
- Interdisciplinary
- Center for Neuroscience Imaging Research (Neuro Technology Group)
- Center for Neuroscience Imaging Research (Cognitive and Computational Neuroscience Group)
- Center for Algorithmic and Robotized Synthesis
- Center for Genome Engineering
- Center for Nanomedicine
- Center for Biomolecular and Cellular Structure
- Center for 2D Quantum Heterostructures
- Center for Quantum Conversion Research
- Institutes
- Korea Virus Research Institute
- News Center
- Career
- Living in Korea
- IBS School





News Center

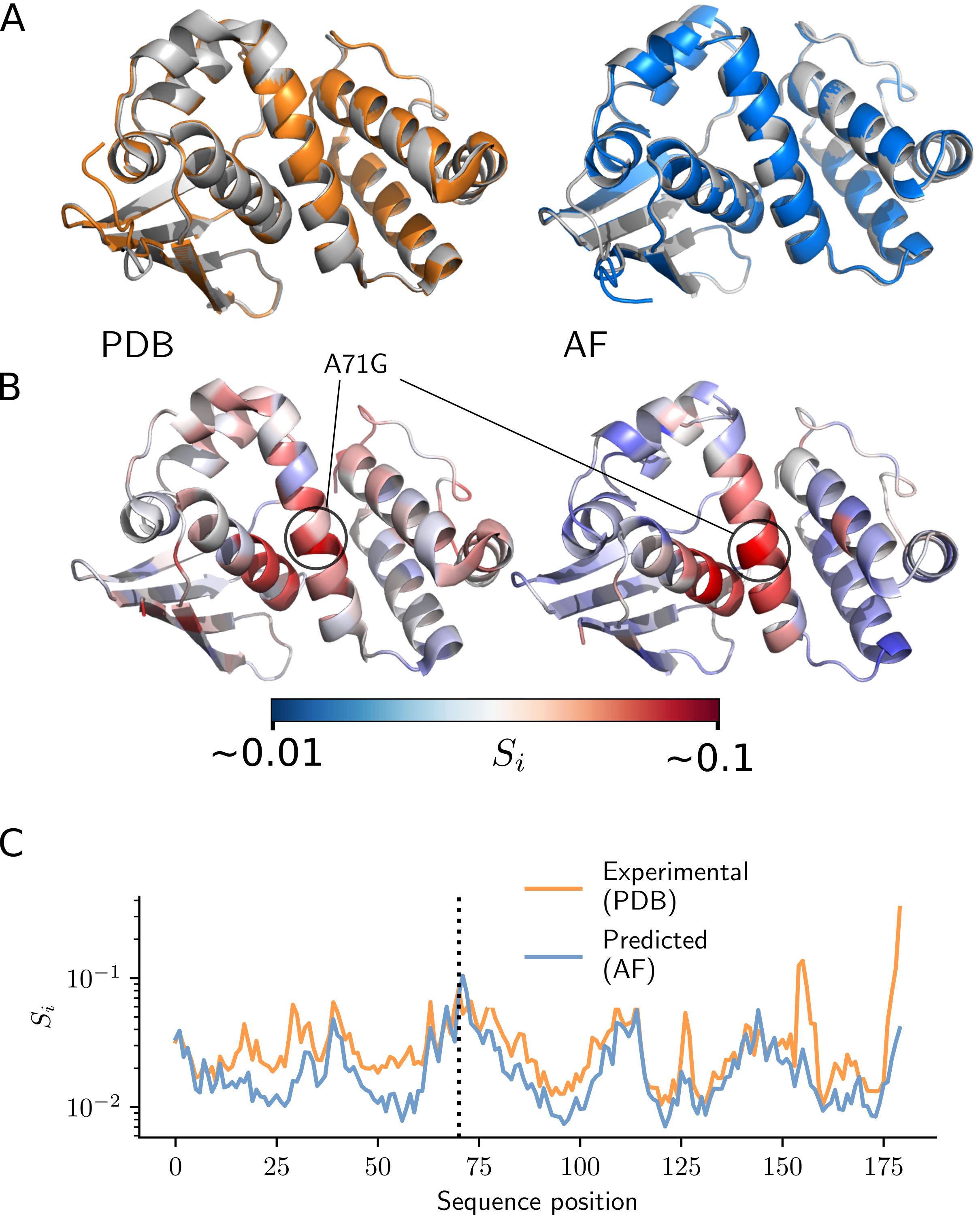
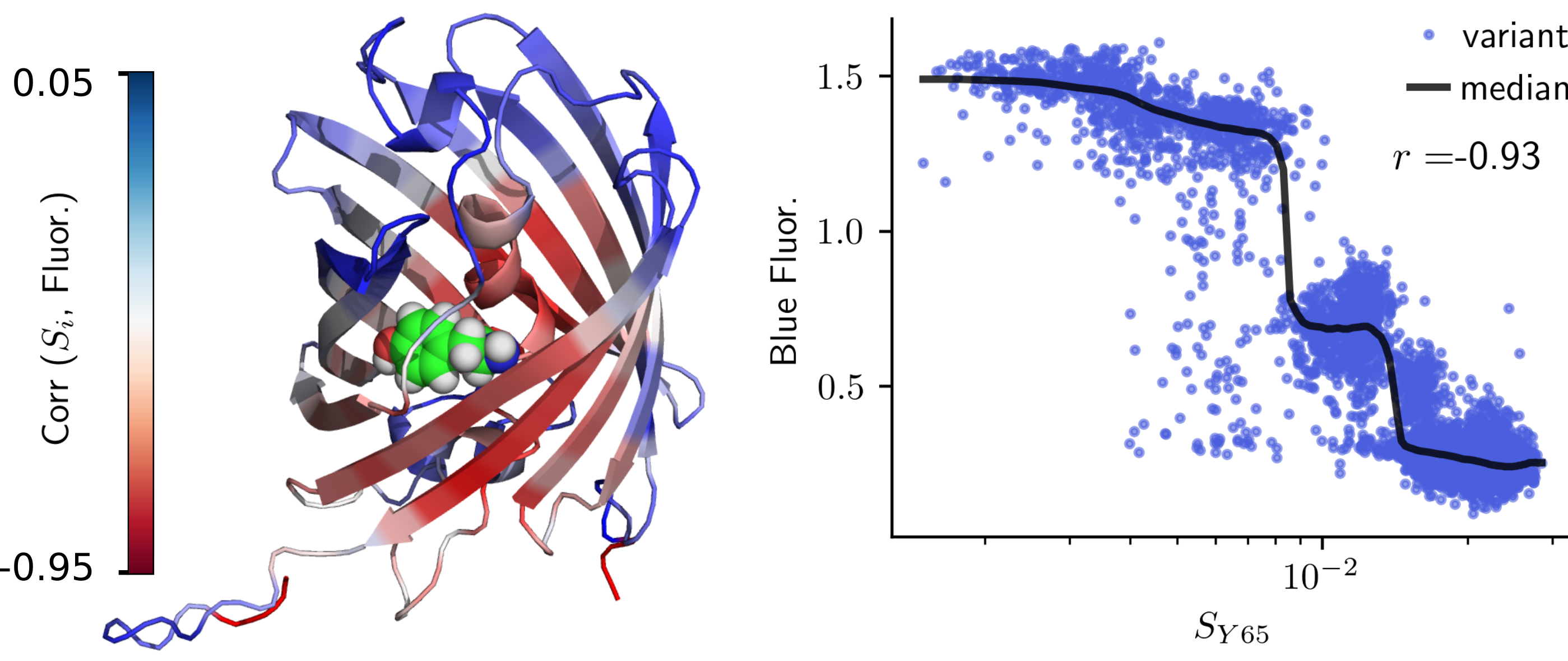
Testing the limits of AlphaFold2’s accuracy in predicting protein structure- AlphaFold2 found to be accurate enough to predict changes in protein structure arising even from single point mutations - Researchers from the Center for Soft and Living Matter within the Institute for Basic Science (South Korea), recently explored the limits of AlphaFold2 AI’s ability to predict protein structures. Proteins, the workhorses of biology, are encoded by DNA sequences and are responsible for vital functions within cells. Since the first experimental measurement of a protein structure was made by John Kendrew in the 1950s, protein’s ability to fold into complex three-dimensional structures has long been a subject of scientific fascination and importance. However, determining these structures experimentally has remained a formidable challenge for decades. IBS researcher John M. McBride said, “Sequencing DNA is a far simpler process than analyzing protein structures. For example, let’s compare the progress of research in DNA and proteins. So far, we have sequenced hundreds of millions of DNA sequences, while on the other hand, we have managed to characterize only several hundred thousand protein structures.” Hence, a central focus of computational biology has been predicting protein structures from their sequences. Understanding a protein's structure is pivotal for deciphering its functions, delving into diseases, unraveling aging, and engineering proteins for various technological applications. Google DeepMind extended the application of artificial intelligence into the biophysics domain. The company’s AlphaFold2 represents the latest milestone in tackling the problem of protein structure prediction, bridging the gap between computational predictions and experimental accuracy. This achievement is substantial enough for some to declare the problem of protein structure prediction as ‘solved.’ But the question is: how accurate is it? Despite AlphaFold2's success in predicting protein structures, questions remain regarding the limits of its accuracy. A fundamental concern arises when attempting to predict the effects of the tiniest changes in a protein – for example, in single point mutation where a single amino acid is substituted with another of differing chemical properties. Achieving accuracy at this level is essential for studying diseases and evolution. There is skepticism about whether AlphaFold2 can achieve such accuracy. The official AlphaFold database clearly states, “AlphaFold has not been validated for predicting the effect of mutations. In particular, AlphaFold is not expected to produce an unfolded protein structure given a sequence containing a destabilizing point mutation.” Additionally, several recent assessments have failed to provide evidence that AlphaFold can predict mutation effects. The IBS researchers used a two-pronged approach to provide a compelling, comprehensive demonstration that AlphaFold can indeed predict mutation effects. First, they directly validated AlphaFold predictions by comparing them with experimental structures. The researchers combined this with an indirect validation of AlphaFold predictions comparing AlphaFold-predicted mutation effects on structure to empirical measurements of protein phenotypes. However, this whole process was extremely challenging. The first major obstacle was that there was very little data that could be used for comparison. Even though there are over half a million structures in the public Protein Data Bank (PDB), only a small fraction of these can be used to measure mutation effects. After rigorous data selection and controlling for various factors, researchers were left with just a few thousand proteins with experimental structures involving minor amino acid changes. These data also contained lots of random noise, which made it challenging to distinguish between structural variations due to measurement error and those caused by mutations. Despite this, the researchers showcased that mutation effects can be statistically measured using experimental structures, providing a robust methodology for quantifying these effects. By applying this methodology, researchers demonstrated that AlphaFold's predictions are nearly as accurate as experimental measurements.The second challenge involves the inadequacy of typical structural similarity measures to capture structural differences due to mutations. Conventional measurements, like the root-mean-square deviation (RMSD), primarily account for changes across the entire protein structure, obscuring small local effects in mutated regions. Local measurements such as the local distance difference test (LDDT) also have low resolution and are limited in their ability to capture fine differences. In response, the research team adopted tools from physics, specifically concepts from continuum mechanics, to measure strain in proteins, a natural measure of deformation. They tested this approach on measurements of fluorescence in several fluorescent proteins. It was found that AlphaFold can accurately predict deformation at the chromophore-binding site (which is important for fluorescence), leading to accurate predictions of fluorescence in fluorescent proteins. Two years after the release of AlphaFold2 we are still exploring the limits and the pitfalls of this fantastic new algorithm. This first successful validation of AlphaFold for predicting mutation effects paves the way for investigations into disease and drug development, leading to improvements in human health. The ability to predict mutation effects will enhance the study of evolution, looking both forward – using directed evolution to develop new enzymes – and backward – understanding the evolutionary history of life itself. The future of protein science is indeed bright.
Notes for editors
- References
- Media Contact
- About the Institute for Basic Science (IBS)
|
| 다음 | |
|---|---|
| 이전 |
- Content Manager
- Public Relations Team : Yim Ji Yeob 042-878-8173
- Last Update 2023-11-28 14:20

55, Expo-ro, Yuseong-gu, Daejeon, Korea, 34126
Tel. +82-42-878-8114 | Fax. +82-42-878-8079 | E-mail. webmaster@ibs.re.kr
Copyright(c) IBS. All Rights Reserved.
