


주메뉴
- About IBS 연구원소개
-
Research Centers
연구단소개
- Research Outcomes
- Mathematics
- Physics
- Center for Underground Physics
- Center for Theoretical Physics of the Universe(Particle Theory and Cosmology Group)
- Center for Theoretical Physics of the Universe(Cosmology, Gravity and Astroparticle Physics Group)
- Center for Exotic Nuclear Studies
- Dark Matter Axion Group
- Center for Artificial Low Dimensional Electronic Systems
- Center for Theoretical Physics of Complex Systems
- Center for Quantum Nanoscience
- Center for Van der Waals Quantum Solids
- Center for Relativistic Laser Science
- Chemistry
- Life Sciences
- Earth Science
- Interdisciplinary
- Center for Neuroscience Imaging Research(Neuro Technology Group)
- Center for Neuroscience Imaging Research(Cognitive and Computational Neuroscience Group)
- Center for Algorithmic and Robotized Synthesis
- Center for Genome Engineering
- Center for Nanomedicine
- Center for Biomolecular and Cellular Structure
- Center for 2D Quantum Heterostructures
- Center for Quantum Conversion Research
- Institutes
- Korea Virus Research Institute
- News Center 뉴스 센터
- Career 인재초빙
- Living in Korea IBS School-UST
- IBS School 윤리경영

주메뉴
- About IBS
-
Research Centers
- Research Outcomes
- Mathematics
- Physics
- Center for Underground Physics
- Center for Theoretical Physics of the Universe(Particle Theory and Cosmology Group)
- Center for Theoretical Physics of the Universe(Cosmology, Gravity and Astroparticle Physics Group)
- Center for Exotic Nuclear Studies
- Dark Matter Axion Group
- Center for Artificial Low Dimensional Electronic Systems
- Center for Theoretical Physics of Complex Systems
- Center for Quantum Nanoscience
- Center for Van der Waals Quantum Solids
- Center for Relativistic Laser Science
- Chemistry
- Life Sciences
- Earth Science
- Interdisciplinary
- Center for Neuroscience Imaging Research(Neuro Technology Group)
- Center for Neuroscience Imaging Research(Cognitive and Computational Neuroscience Group)
- Center for Algorithmic and Robotized Synthesis
- Center for Genome Engineering
- Center for Nanomedicine
- Center for Biomolecular and Cellular Structure
- Center for 2D Quantum Heterostructures
- Center for Quantum Conversion Research
- Institutes
- Korea Virus Research Institute
- News Center
- Career
- Living in Korea
- IBS School





News Center

|
Identifying a mechanism for social deficit in autism spectrum disorders The IBS Center for Synaptic Brain Dysfunctions (Director Eunjoon Kim) found that mice lacking the excitatory synaptic signaling scaffold IRSp53 showed social deficit and enhanced NMDA receptor function in the hippocampus. Normalization of NMDA receptor function in these mice by drugs rescued social impairment, suggesting that deviation of NMDAR function leads to social deficits and that correcting the deviation has beneficial effects. Social interactions are the pillars of human
activity, with institutions such as marriage,
family, and friendships immortalized in all
manners of art and literature, and thus upheld
as being the pinnacles of a good life. We all
feel these interactions through the warmth of
a mother’s love, the strength of the friend’s
support, or the passion in a lover’s embrace.
When a person lacks the ability to show such
emotions, society finds it hard to accept them.
However, such is the fate of children who, by
no fault of their own, are born with autism
spectrum disorders (ASDs).
ASDs, commonly referred to as autism, is
defined by two core symptoms: (a) deficits
in social communication and (b) restricted,
repetitive patterns of behavior. The Center for
Disease Control (CDC, USA) reported that the
prevalence of autism was 1 in 68 children, or
about 1.5%. You can rest assured that 1 in 68
families whole are also affected deeply by this
disorder. The need to understand and overcome
this disorder increases with each child diagnosed
and with each family struck to its core by this
debilitating condition.
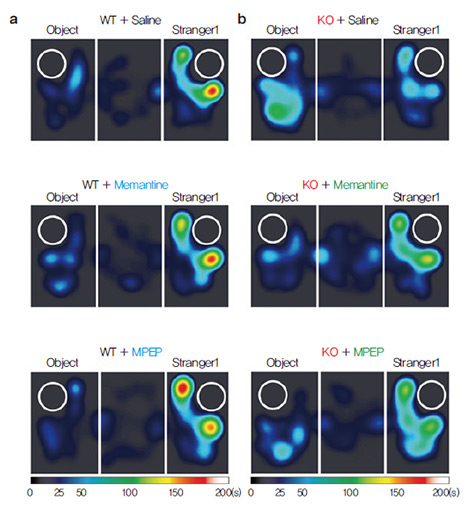
a. Wild-type (WT) mice show Stranger1
far greater interest in other mice
(Stranger 1) than inanimate
objects (Object), as depicted in
the heat map. It turns out that these mice displayed NMDAR
hyper-function at the synapse. A NMDA
receptor (NMDAR) is a type of ion channel,
present in the synapse and heavily influences
the efficiency of synaptic transmission during
neuronal activity. When the research group
treated the mice with a drug called memantine,
which reduces the activity of NMDAR, the
same mice reverted to normal levels of social
interaction, comparable to that of wildtype
mice. As memantine directly lodges
into the channel of NMDAR, the research
group tried to elicit the same rescue effect by targeting NMDAR indirectly via the mGluR5
pathway with a drug called MPEP, in order
to confirm the hypothesis in a secondary
manner. The rescuing effect of the drugs were
confirmed at the synapse level as well, where
electrophysiological recordings of synapse
activity showed a normalization of NMDAR
activity once applied with either drugs. Published paper |
| before | |
|---|---|
| Next |
- Content Manager
- Public Relations Team : Yim Ji Yeob 042-878-8173
- Last Update 2023-11-28 14:20

55, Expo-ro, Yuseong-gu, Daejeon, Korea, 34126
Tel. +82-42-878-8114 | Fax. +82-42-878-8079 | E-mail. webmaster@ibs.re.kr
Copyright(c) IBS. All Rights Reserved.
