


주메뉴
- About IBS 연구원소개
-
Research Centers
연구단소개
- Research Outcomes
- Mathematics
- Physics
- Center for Theoretical Physics of the Universe(Particle Theory and Cosmology Group)
- Center for Theoretical Physics of the Universe(Cosmology, Gravity and Astroparticle Physics Group)
- Center for Exotic Nuclear Studies
- Center for Artificial Low Dimensional Electronic Systems
- Center for Underground Physics
- Center for Axion and Precision Physics Research
- Center for Theoretical Physics of Complex Systems
- Center for Quantum Nanoscience
- Center for Van der Waals Quantum Solids
- Chemistry
- Life Sciences
- Earth Science
- Interdisciplinary
- Institutes
- Korea Virus Research Institute
- News Center 뉴스 센터
- Career 인재초빙
- Living in Korea IBS School-UST
- IBS School 윤리경영

주메뉴
- About IBS
-
Research Centers
- Research Outcomes
- Mathematics
- Physics
- Center for Theoretical Physics of the Universe(Particle Theory and Cosmology Group)
- Center for Theoretical Physics of the Universe(Cosmology, Gravity and Astroparticle Physics Group)
- Center for Exotic Nuclear Studies
- Center for Artificial Low Dimensional Electronic Systems
- Center for Underground Physics
- Center for Axion and Precision Physics Research
- Center for Theoretical Physics of Complex Systems
- Center for Quantum Nanoscience
- Center for Van der Waals Quantum Solids
- Chemistry
- Life Sciences
- Earth Science
- Interdisciplinary
- Institutes
- Korea Virus Research Institute
- News Center
- Career
- Living in Korea
- IBS School





News Center

How to Code a Functional Molecular Machine?- Researchers have designed a model to explain how evolution can shape flexible, functional proteins.- An international team has developed a model that simulates protein evolution. Starting from stiff, unfunctional proteins, the computer model shows how evolving protein components can work together to give rise to dynamic and efficient molecular machines. Flexibility allows proteins to change their 3D conformation to bind other molecules: this property is crucial to their function. Prof. Tsvi Tlusty and Dr. Sandipan Dutta at the Center for Soft and Living Matter, within the Institute for Basic Science (IBS, South Korea), in collaboration with Prof. Albert Libchaber from Rockefeller University and Prof. Jean-Pierre Eckmann from University of Geneva have mimicked gene evolution to obtain proteins that can bend and bind to other molecules. Understanding of this relation is one of the most highly sought-after aspects of protein biology; it could help to explain pharmaceutical action of drugs binding to their targets. Evolution has shaped the living world we see around us for billions of years. Zillions of proteins work harmoniously to keep these life processes going. They are responsible for the smooth functioning of any organism: they recognize other molecules (ligands), bind to them and convert them. Others have transport function, provide structure, and support to the cells. Genes store the information about the production and design of these molecular machines. However, despite decades of research, drafting the “map” that draws the path from genes to protein function is not trivial. According to a recent hypothesis, protein function relies on “flexible joints.” This study, published in Proceedings of the National Academy of Sciences (PNAS), examines the link between function and flexibility by modelling proteins like elastic networks. In this model, proteins are made of flexible (polar) and rigid (hydrophobic) amino acids connected by molecular “springs”. If some regions of the protein are flexible enough, they form a “floppy” channel, and the entire molecular machine can bend like a hinge. This motion allows them to bind effectively to other molecules. The binding between a ligand and a stiff or flexible protein can be thought as a ball landing on a rock or a soft pillow. The ball is likely to bounce away after hitting the rock, but the pillow is more likely to accept it. Therefore, the flexible protein is a better binder. In this model, genes store the details of the protein design in a binary fashion: flexible amino acids are stored as zeros and rigid amino acids as ones. As a result, the entire protein structure can be simplified as a code, like 11110001...111, similar to the digital memory of a computer. However, not all codes give rise to functional proteins, for example a code with only ones: 111111…1111, would give rise to an entirely stiff protein, unable to move, and nonfunctional. Among all possible codes, only some produce a functional protein with a “floppy” region in the center that can welcome the ligand. The model mimics evolution by changing one random amino acid at a time. During evolution, the zeros and ones in the gene are randomly flipped through a process called mutation. Most mutations do not bring any difference, or lead to non-functional proteins, but some rare mutations can give rise to a more efficient protein. Essentially, both functional and non-functional proteins are produced during evolution, but according to Darwin’s theory of “survival of the fittest”, only the functional proteins are kept and the non-functional proteins eventually die out.
What does a “functional” code look like? The answer is not straightforward. In fact, the number of codes of a functional protein, even a simple protein, is enormous, larger than the size of the universe. However, using techniques of data analysis, it is possible to search for hidden patterns in all functional codes to look for some unifying characteristics. For example, the “floppy” channel in the protein has interesting and peculiar features, and a mutation at one end of the channel has long-range effects which can strongly affect the maintenance of mutations of other distant amino acids. “In the future, we plan to explore how to apply this study to real proteins, like kinases, ” said Group Leader Tsvi Tlusty, a correspondent in the study. “Moreover, the study opens avenues to investigate the evolution of other protein functions, like molecular recognition. Using huge databases, which have been developed through years of research, can probably uncover some underlying phenomena on the evolution of proteins.” Burdette Choi, Letizia Diamante Notes for editors - References - Media Contact - About the Institute for Basic Science (IBS) |
|||
Center for Soft and Living MatterPublication Repository |
|||
|
|
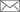
| Next | |
|---|---|
| before |
- Content Manager
- Public Relations Team : Suh, William Insang 042-878-8137
- Last Update 2023-11-28 14:20

55, Expo-ro, Yuseong-gu, Daejeon, Korea, 34126
Tel. +82-42-878-8114 | Fax. +82-42-878-8079 | E-mail. webmaster@ibs.re.kr
Copyright(c) IBS. All Rights Reserved.
